About
Opqua is an epidemiological modeling framework for pathogen population genetics and evolution.
Opqua stochastically simulates pathogens with distinct, evolving genotypes that spread through populations of hosts which can have specific immune profiles.
Opqua is a useful tool to test out scenarios, explore hypotheses, make predictions, and teach about the relationship between pathogen evolution and epidemiology.
Among other things, Opqua can model
host-host, vector-borne, and vertical transmission
pathogen evolution through mutation, recombination, and/or reassortment
host recovery, death, and birth
metapopulations with complex structure and demographic interactions
interventions and events altering demographic, ecological, or evolutionary parameters
treatment and immunization of hosts or vectors
influence of pathogen genome sequences on transmission and evolution, as well as host demographic dynamics
intra- and inter-host competition and evolution of pathogen strains across user-specified adaptive landscapes
How Does Opqua Work?
Basic concepts
Opqua models are composed of populations containing hosts and/or vectors, which themselves may be infected by a number of pathogens with different genomes.
A genome is represented as a string of characters. All genomes must be of the same length (a set number of loci), and each position within the genome can have one of a number of different characters specified by the user (corresponding to different alleles). Different loci in the genome may have different possible alleles available to them. Genomes may be composed of separate chromosomes, separated by the “/” character, which is reserved for this purpose.
Each population may have its own unique parameters dictating the events that happen inside of it, including how pathogens are spread between its hosts and vectors.
Events
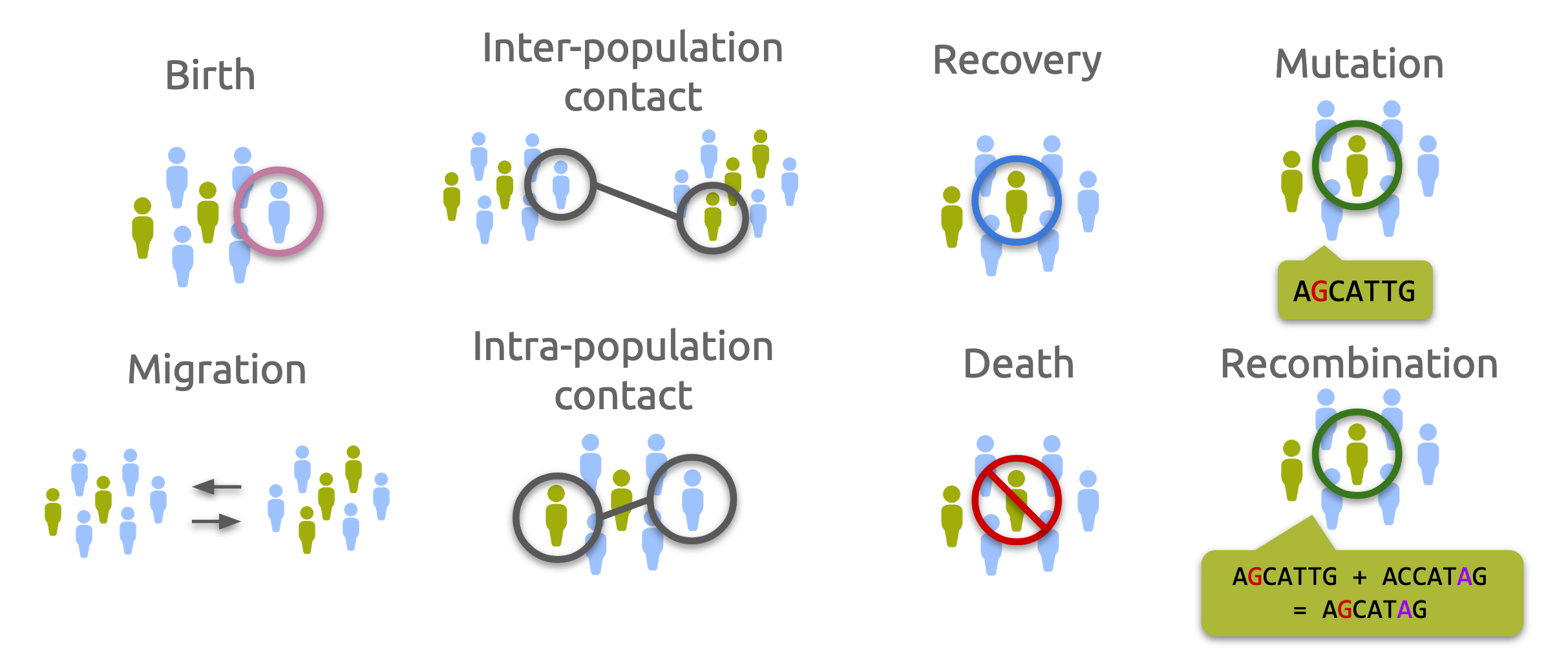
There are different kinds of events that may occur to hosts and vectors in a population:
contact between an infectious host/vector and another host/vector in the same population (intra-population contact) or in a different population (“population contact”)
migration of a host/vector from one population to another
recovery of an infected host/vector
birth of a new host/vector from an existing host/vector
death of a host/vector due to pathogen infection or by “natural” causes
mutation of a pathogen in an infected host/vector
recombination of two pathogens in an infected host/vector
The likelihood of each event occurring is determined by the population’s
parameters (explained in the newSetup() function documentation) and
the number of infected and healthy hosts and/or vectors in the population(s)
involved. Crucially, it is also determined by the genome sequences of the
pathogens infecting those hosts and vectors. The user may specify arbitrary
functions to evaluate how a genome sequence affects any of the above kinds of
rates. This is once again done through arguments of the newSetup()
function. As an example, a specific genome sequence may result in increased
transmission within populations but decreased migration of infected hosts, or
increased mutation rates. These custom functions may be different across
populations, resulting in different adaptive landscapes within different
populations.
Contacts within and between populations may happen by any combination of host-host, host-vector, and/or vector-host routes, depending on the populations’ parameters. When a contact occurs, each pathogen genome present in the infecting host/vector may be transferred to the receiving host/vector as long as one “infectious unit” is inoculated. The number of infectious units inoculated is randomly distributed based on a Poisson probability distribution. The mean of this distribution is set by the receiving host/vector’s population parameters, and is multiplied by the fraction of total intra-host fitness of each pathogen genome. For instance, consider the mean inoculum size for a host in a given population is 10 units and the infecting host/vector has two pathogens with fitnesses of 0.3 and 0.7, respectively. This would make the means of the Poisson distributions used to generate random infections for each pathogen equal to 3 and 7, respectively.
Inter-population contacts occur via the same mechanism as intra-population contacts, with the distinction that the two populations must be linked in a compatible way. As an example, if a vector-borne model with two separate populations is to allow vectors from Population A to contact hosts in Population B, then the contact rate of vectors in Population A and the contact rate of hosts in Population B must both be greater than zero. Migration of hosts/vectors from one population to another depends on a single rate defining the frequency of vector/host transport events from a given population to another. Therefore, Population A would have a specific migration rate dictating transport to Population B, and Population B would have a separate rate governing transport towards A.
Recovery of an infected host or vector results in all pathogens being removed from the host/vector. Additionally, the host/vector may optionally gain protection from pathogens that contain specific genome sequences present in the genomes of the pathogens it recovered from, representing immune memory. The user may specify a population parameter delimiting the contiguous loci in the genome that are saved on the recovered host/vector as “protection sequences”. Pathogens containing any of the host/vector’s protection sequences will not be able to infect the host/vector.
Births result in a new host/vector that may optionally inherit its parent’s protection sequences. Additionally, a parent may optionally infect its offspring at birth following a Poisson sampling process equivalent to the one described for other contact events above. Deaths of existing hosts/vectors can occur both naturally or due to infection mortality. Only deaths due to infection are tracked and recorded in the model’s history.
De novo mutation of a pathogen in a given host/vector results in a single locus within a pathogen’s genome being randomly assigned a new allele from the possible alleles at that position. Recombination of two pathogens in a given host/vector creates two new genomes based on the independent segregation of chromosomes (or reassortment of genome segments, depending on the field) from the two parent genomes. In addition, there may be a Poisson-distributed random number of crossover events between homologous parent chromosomes. Recombination by crossover event will result in all the loci in the chromosome on one side of the crossover event location being inherited from one of the parents, while the remainder of the chromosome is inherited from the other parent. The locations of crossover events are distributed throughout the genome following a uniform random distribution.
Interventions
Furthermore, the user may specify changes in model behavior at specific timepoints during the simulation. These changes are known as “interventions”. Interventions can include any kind of manipulation to populations in the model, including:
adding new populations
changing a population’s event parameters and adaptive landscape functions
linking and unlinking populations through migration or inter-population contact
adding and removing hosts and vectors to a population
Interventions can also include actions that involve specific hosts or vectors in a given population, such as:
adding pathogens with specific genomes to a host/vector
removing all protection sequences from some hosts/vectors in a population
applying a “treatment” in a population that cures some of its hosts/vectors of pathogens
applying a “vaccine” in a population that protects some of its hosts/vectors from pathogens
For these kinds of interventions involving specific pathogens in a population, the user may choose to apply them to a randomly-sampled fraction of hosts/vectors in a population, or to a specific group of individuals. This is useful when simulating consecutive interventions on the same specific group within a population. A single model may contain multiple groups of individuals and the same individual may be a member of multiple different groups. Individuals remain in the same group even if they migrate away from the population they were chosen in.
When a host/vector is given a “treatment”, it removes all pathogens within the host/vector that do not contain a collection of “resistance sequences”. A treatment may have multiple resistance sequences. A pathogen must contain all of these within its genome in order to avoid being removed. On the other hand, applying a vaccine consists of adding a specific protection sequence to hosts/vectors, which behaves as explained above for recovered hosts/vectors when they acquire immune protection, if the model allows it.
Simulation
Models are simulated using an implementation of the Gillespie algorithm in which the rates of different kinds of events across different populations are computed with each population’s parameters and current state, and are then stored in a matrix. In addition, each population has host and vector matrices containing coefficients that represent the contribution of each host and vector, respectively, to the rates in the master model rate matrix. Each coefficient is dependent on the genomes of the pathogens infecting its corresponding vector or host. Whenever an event occurs, the corresponding entries in the population matrix are updated, and the master rate matrix is recomputed based on this information.

The model’s state at any given time comprises all populations, their hosts and vectors, and the pathogen genomes infecting each of these. A copy of the model’s state is saved at every time point, or at intermittent intervals throughout the course of the simulation. A random sample of hosts and/or vectors may be saved instead of the entire model as a means of reducing memory footprint.
Output
The output of a model can be saved in multiple ways. The model state at each saved timepoint may be output in a single, raw pandas DataFrame, and saved as a tabular file. Other data output types include counts of pathogen genomes or protection sequences for the model, as well as time of first emergence for each pathogen genome and genome distance matrices for every timepoint sampled. The user can also create different kinds of plots to visualize the results. These include:
plots of the number of hosts and/or vectors in different epidemiological compartments (naive, infected, recovered, and dead) across simulation time
plots of the number of individuals in a compartment for different populations
plots of the genomic composition of the pathogen population over time
phylogenies of pathogen genomes
Users can also use the data output formats to make their own custom plots.